蛋白质紊乱和聚集在许多神经退行性疾病的发病机制中发挥着重要作用,例如阿尔茨海默病和帕金森病。这些疾病中聚集过程的最终产物是富含β-折叠的淀粉样蛋白原纤维。尽管在大多数情况下,淀粉样蛋白聚集过程中形成的可溶性小低聚物是有毒物质。为了揭示各种淀粉样蛋白疾病的分子基础,则需要全面了解蛋白质聚集过程背后的物理化学力。在用于研究蛋白质聚集的众多生物物理和生化技术中,原子水平的(MD)模拟提供了该过程的最高时间和空间分辨率,捕获了淀粉样蛋白低聚物形成过程中的关键步骤。在此作者提供了使用GROMACS设置、运行和分析聚合肽的MD模拟的各个步骤的操作指南。对于结果分析,作者也提供了其实验室开发的脚本,可以确定驱动聚合过程的寡聚体大小和肽间接触。此外,作者也提供了从肽聚集的MD数据中导出马尔可夫状态模型和转换网络的工具。
在蛋白质聚集过程中,错误折叠或本质上无序的蛋白质首先组装成寡聚体,寡聚体可以长成高度有序的β-折叠聚集体,称为淀粉样蛋白原纤维,该过程可以发生在细胞内或细胞外环境,并且与各种神经退行性疾病高度相关,例如阿尔茨海默病和帕金森病。神经退行性疾病是导致神经细胞进行性退化死亡的退化性疾病,可导致运动功能(称为共济失调)或精神功能(称为痴呆)问题。目前,上述与淀粉样蛋白聚集有关的疾病均无法治愈。
计算机模拟,尤其是分子动力学(MD)模拟已成为研究蛋白质构象和结构特性的重要工具。进入21世纪,强大的超级计算机使我们能够模拟越来越多的复杂系统,用于更长的时间尺度和更大的长度尺度,从而更加接近真实的实验条件。
另外,MD模拟会产生大量数据,从中提取有关分子过程的信息需要复杂的后处理技术。其中一种被称为马尔可夫状态模型(MSM)最近在计算生物化学和生物物理学领域变得越来越重要。作为一种用于阐明隐藏在MD数据中的相关状态和过程的技术,MSM是以状态和速率格式对系统动力学进行编码的网络模型,即分子系统可以在特定时间点以多种可能状态之一存在。在特定的时间间隔内,具有转换到其他状态的固定概率,包括其本身。MSM的一个基本假设是无记忆性,即从一种状态转换到另一种状态的概率仅取决于当前状态,而不取决于系统的历史。利用MSM从MD数据中提取基本信息已被证明适用于各种生物系统,包括蛋白质折叠、蛋白质-配体结合或变构以及分子自组装等过程。这种方法在阐明动力学相关聚集途径方面也已被证明可用于淀粉样肽KFFE的自组装过程。
另外,由Strodel实验室开发的过渡网络(TN)则提供了另一种表征蛋白质聚集的网络模型。TNs基于构象聚类,而不是MSM 中所做的动力学聚类。在TN中,聚合状态是根据与描述所研究的聚合过程最相关的特征来定义的。这些所谓的描述符总是包括聚集体的大小,并通过聚集体中蛋白质之间相互作用的数量和类型、它们的形状和β-折叠的量(即与淀粉样蛋白聚集相关的量)来增强。将高维构象空间转换到这个低维TN空间可以清楚地看到聚集过程的结构和路径。该方法已被成功应用于与阿尔茨海默病的发展相关的淀粉样蛋白-β肽(Aβ42)的聚集以及自酵母朊病毒蛋白Sup35的GNNQQNY极性肽序列的研究。
本研究示例了一种执行蛋白质聚集的MD模拟研究,并利用马尔可夫状态模型或过渡网络对其模拟数据进行了深度的统计分析。
模拟所需工具
本教程以GROMACS-2016.4为MD工具,Charmm36m为蛋白力场,TIP3P水模型,模拟了6份Aβ16-22蛋白质的聚集情况。使用Charmm36m力场的原因是因为Charmm36m力场已被证明是模拟Aβ16-22的最佳力场之一。完成本次模拟需要的工具和软件包有:
1) 蛋白质可视化程序,如PyMOL或VMD;
2) 用于一般数据分析的Python;
3) 专门设计用于分析MD轨迹的Python库,如MDAnalysis和MDTraj;
4) 分子包装优化软件,如PACKMOL;
5 ) Gromacs分子动力学软件包;
初始结构准备
本教程以Aβ16-22为例,在两端添加基团进行封端处理,从而有序列ACE-KLVFFAE-NME。由于蛋白质数据库(Protein DataBank,PDB)不包含该肽的结构,因此可以从Aβ42的一个PDB结构的残基16- 21的坐标中检索出如下模拟的起始结构(PDBID:1Z0Q)。利用PyMOLProtein模式下的Builder工具,可以分别在残基16的N端和残基22的C端加入ACE和NME基团。
包含6条随机肽段的水盒子的构建
第一步是为Aβ16-22单体产生松弛构象。模拟长度取决于所研究多肽的大小,对于Aβ16-22,建议模拟长度为1μs或更长。其中,可以使用构象聚类来获得最稳定的单体结构。随机将六个Aβ16-22单体放置在模拟盒中作为初始系统。在此之前需要进行单体的初始模拟从而避免在后续步骤中产生可能的聚集等非自然状态的结构,这会花费更多模拟时间来松弛这些聚集体,或导致模拟失败。
构建初始体系方法一:使用PACKMOL软件将六条肽段随机分布在边长为10nm的正方体盒子内,每条肽段距离至少12埃(1.2nm)。PACKMOL运行命令
packmol< packmol.inp
参数文件packmol.inp内容:
#Sixmonomers of abeta16-22 peptide
#minimum distance between twomonomers
tolerance 12.0
seed -1
#The file type ofinput and output files is PDB
filetype pdb
#The name of theoutput file
output abeta16-22_hexamer.pdb
#add TER to aftereach monomer chain
add_amber_ter
#distance from theedges of box
add_box_sides 1.0
#path to input structurefile
#units for distance is measured in Angstrom
#box sizeis 100 Å
structure abeta16-22.pdb
number 6
insidebox 0. 0. 0. 100. 100. 100.
end structure
构建初始体系方法二:使用Gromacs自带的insert命令完成:
gmxinsert -molecules -ci abeta16-22.pdb -nmol 6 -box 10 10 10 -oabeta16-22_hexamer.pdb
构建完成后,打开abeta16-22_hexamer.pdb,有类似下面的六条随机分布的肽段。


六个Aβ16-22肽(显示为不同颜色的表面)的说明性示例,随机放置在被溶剂分子包围的立方体中(显示为灰点)。
构建模拟所需文件目录和参数文件
为了避免替换文件,建议在各自不同的目录中执行相应的模拟步骤。这里创建五个主要步骤的目录:拓扑构建、能量最小化、NVT均衡、NPT均衡和MD模拟运行。
mkdir1-topol 2-em 3-nvt 4-npt 5-md
对于每一步都需要一个mdp文件。mdp文件类型设置了分子动力学参数,因为这些文件包含了GROMACS建立MD模拟的所有关键参数。所需的5个mdp文件如下所示,将所有的mdp文件分别放在新建的mdp文件夹下。
mkdir mdp
“ions.mdp”for adding ions
;;ions.mdp ; Run setup integrator = steep emtol = 1000 emstep = 0.01nsteps = 2000 ; Neighbor search cutoff-scheme = Verlet pbc = xyz ;Electrostatics and vdW coulombtype = PME pme-order = 4 fourierspacing= 0.1 rcoulomb = 1.2 rvdw = 1.2
“em.mdp”for energy minimization
;;em.mdp ; Run setup integrator = steep emtol = 500 emstep = 0.001nsteps = 2000 nstxout = 100 ; Neighbor searching cutoff-scheme =Verlet nstlist = 20 ns-type = grid pbc = xyz ; Electrostaticscoulombtype = PME pme-order = 4 fourierspacing = 0.1 rcoulomb = 1.2 ;VdW rvdw = 1.2
“nvt.mdp”for first equilibration in the NVT ensemble
;;nvt.mdp define = -DPOSRES ; Run setup integrator = md dt = 0.002 ; 2fs nsteps = 50000 ; Output control nstxout = 5000 nstvout = 5000nstfout = 5000 nstlog = 500 nstenergy = 500 ; Bonds constraints =all-bonds constraint-algorithm = LINCS lincs-order = 4 lincs-iter = 1; Neighbor searching cutoff-scheme = Verlet nstlist = 20 ns-type =grid pbc = xyz ; Electrostatics coulombtype = PME pme-order = 4fourierspacing = 0.1 rcoulomb = 1.2 ; VdW rvdw = 1.2 ; T coupling ison tcoupl = v-rescale tc-grps = Protein Non-Protein tau-t = 0.1 0.1ref-t = 300 300 nsttcouple = 10 ; P coupling is off pcoupl = no ;Velocity generation gen-vel = yes gen-temp = 300 continuation = no
“npt.mdp”for second equilibration in the NPT ensemble
;;npt.mdp define = -DPOSRES ; Run setup integrator = md dt = 0.002nsteps = 100000 ; Output control nstxout = 5000 nstvout = 5000nstfout = 5000 nstlog = 500 nstenergy = 500 ; Bonds constraints =all-bonds constraint-algorithm = LINCS
“md.mdp”for MD production run
;;md.mdp ; Run setup integrator = md dt = 0.002 nsteps = 500000000 ;500 million ; Output control nstxout = 0 nstvout = 0 nstfout = 0nstlog = 2500 nstenergy = 2500 nstxout-compressed = 2500compressed-x-grps = System ; Bonds constraints = all-bondsconstraint-algorithm = LINCS lincs-order = 4 ; Neighbor searchingcutoff-scheme = Verlet Nstlist = 20 ns-type = grid pbc = xyz ;Electrostatics coulombtype = PME pme-order = 4 fourierspacing = 0.1rcoulomb = 1.2 ; VdW rvdw = 1.2 ; T coupling is on tcoupl = v-rescaletc-grps = Protein Non-Protein tau-t = 0.1 0.1 ref-t = 300 300nsttcouple = 10 ; Pressure coupling is off pcoupl = Parrinello-Rahmanpcoupltype = isotropic tau_p = 2.0 ref_p = 1.0 compressibility =4.5e-5 ; Velocity generation gen-vel = no gen-temp = 300 continuation= yes
构建拓扑文件
这一步中需要创建模拟用的拓扑输入文件。该文件包含了关于分子类型和分子数目信息。除拓扑文件外,还生成一个gro文件,该文件与pdb文件一样,也包含模拟系统的坐标,只是格式有区别。另外gro文件也可以记录原子速度。由于Gromacs不自带Charmm36力场,需要自行下载,并放在topol目录下,下载地址:
http://mackerell.umaryland.edu/download.php?filename=CHARMM_ff_params_files/charmm36-mar2019.ff.tgz
然后使用Gromacs的pdb2gmx命令生成拓扑文件、itp和gro文件。命令如下:
gmxpdb2gmx -f ../abeta16-22_hexamer.pdb -o protein.gro -p topol.top-ignh -ter <<EOF
1
1
3
4
3
4
3
4
3
4
3
4
3
4
EOF
参数详解:
-f:读取输入结构文件abeta16-22_hexamer. pdb
-o和-p:编写输出结构文件Protein.gro和系统拓扑文件topol.top
-ignh:忽略输入文件中的氢原子,这是可取的,因为输入文件和力场中氢原子的命名约定不同。新的氢原子会被加入GROMACS使用选定力场的H原子名称。
-ter:为N端和C端交互分配电荷状态
方案1:选择蛋白质力场(charmm36-mar2019 )。
方案1:选择水力场(TIP3P )
方案3:选择N端(无,如我们使用ACE复盖)。
方案4:选择C终点(没有,因为我们使用NME复盖)。
对于系统中的每个肽段,选项3和4重复,本案例中重复6次。
生成盒子
接下来生成放置肽段的盒子和填充TIP3P水分子
gmxeditconf -f protein.gro -o box.gro -bt cubic -box 10 10 10gmx solvate-cp box.gro -cs spc216.gro -o protein-solvated.gro -p topol.top
填充溶剂
模拟是在周期性边界条件下进行,因此需要对体系进行电中和,这里使用150mM的NaCl进行填充体系。
gmxgrompp -f ../mdp/ions.mdp -c protein-solvated.gro -p topol.top -oprotein-ions.tprecho 13 | gmx genion -s protein-ions.tpr -oprotein-ions.gro -p neutral -conc 0.15
参数详解:
-neutral:使用Na+和Cl-中和体系
-conc0.15:150 mM的NaCl进行填充体系
echo13,使用上述离子替换体系中的水分子
能量最小化
进入到2-em文件夹,使用如下命令进行体系的能量最小化
gmxgrompp -f ../mdp/em.mdp -c ../1-topol/protein-ions.gro -p../1-topol/topol.top -o protein-em.tprgmx mdrun -v -deffnm protein-em
命令运行成功后会生成:能量最小化后的结构文件protein-em.gro、能量信息记录文件protein-em.edr和轨迹文件protein-em.trr。
NVP和NP平衡、MD模拟
在EM步骤之后,进行两个平衡(EQ )步骤。第一步EQ是在等温线和等温条件下进行的,当N(粒子数),V(体积)和T(温度)保持不变时,称为NVT系综。而且,在这一步中,只有溶剂分子和离子在多肽周围达到平衡,使其达到预期的温度(在本案例中,温度为300K),而多肽原子的位置固定不变。为此,使用在拓扑构建过程中生成的包含位置约束的.itp文件。
cd ../3-nvt
gmxgrompp -f ../mdp/nvt.mdp -c ../1-topol/protein-em.gro -p../1-topol/topol.top -o protein-nvt.tpr
gmx mdrun -v -deffnmprotein-nvt
在第2个EQ阶段,利用等温等压系综(又称NPT系综)调节系统的压力和密度,使用grompp和mdrun命令执行200ps的NPT模拟:
cd ../4-npt
gmxgrompp -f ../mdp/npt.mdp -c ../1-topol/protein-nvt.gro -p../1-topol/topol.top -o protein-npt.tpr
gmx mdrun -v -deffnmprotein-npt
在完成EM和2个EQ后,就可以进行最终正式的动力学模拟了。该模拟去除了对蛋白质的位置限制,但使用LINCS方法将所有键长限制在它们的平衡值,该方法允许使用较短的2fs的时间步长来积分运动方程。为了充分采样构象空间,总的模拟时间需要以微秒级别运行。
本例中执行1-μsMD模拟,首先切换到相应目录,调用GROMACS 预处理器grompp,然后调用mdrun 命令:
cd ../5-md
gmxgrompp -f ../mdp/md.mdp -c ../1-topol/protein-npt.gro -p../1-topol/topol.top -o protein-md.tpr
gmx mdrun -v -deffnmprotein-md
在MD运行完成之后,将生成xtc文件,一种使用较低精度保存的轨迹可移植格式文件。
结果分析
protein_md.xtc文件包含用于分析模拟采样系统的所有坐标。GROMACS自带的分析工具可以读取xtc文件的二进制格式。但在本文中,作者使用基于Python的MDAnalysis 和类似MDTraj的工具,它们也可以处理xtc文件,并与内部的Python 脚本相关联。对于具有六个 Aβ16-22肽的系统的当前示例,我们将分析寡聚体的大小和聚集体中的肽间接触。首先我们新建analysis目录,进入该目录cd ../ mkdir analysis cd analysis/
cd ../
mkdiranalysis
cd analysis/
将MD目录下轨迹文件protein_md.xtc以及输入文件 protein_md.tpr 拷贝进该analysis目录下。
对于分析来说,我们只需要蛋白的坐标而不需要溶剂和离子的坐标。使用如下命令提取和重新保存该文件
gmxtrjconv -s protein_md.tpr -f protein_md.xtc -o protein_only.xtc
随后选择“1”将蛋白处于坐标中心,然后选择“1”输出蛋白坐标到xtc文件中。
我们还需要提取 .gro 或 .pdb 格式的参考结构文件。在当前示例中,我们创建一个 .pdb 文件。 gmx trjconv -s protein_md.tpr -f protein_md.xtc -o protein_only.pdb -dump 0 在命令提示符下:为输出“protein”选择选项“1” (解释: -dump 0:转储轨迹文件的第一帧)。
蛋白质聚集模拟存在一个特殊问题需要在分析之前解决,即 MD 模拟过程中存在的周期性盒子可能会导致蛋白质看起来被破坏或者截断。许多分析脚本无法处理此类损坏的蛋白质,这会导致分析中出现伪影。为此我们使用 VMD读取新创建的 protein_only.pdb 和 protein_only.xtc 文件并可视化轨迹。然后在 Extensions 选项卡上打开 Tk Console 并通过输入以下命令可视化蛋白质系统周围的 PBC 框:pbc box
在播放轨迹电影时,可以看到一些帧的分子破碎,可以在Tk控制台中输入以下命令重新组装:
[atomselecttop all] set chain 0 pbc join fragment -all
之后,将所有坐标保存为分析目录中的新名称protein_nopbc.trr。这个轨迹文件将用于下面的分析。
或者使用gmx自带命令进行PBC处理,去除周期性边界:
gmxtrjconv -s protein_only.pdb -f protein_only.xtc -pbc nojump -oprotein_nopbc.trr
用于计算寡聚化状态(单体、二聚体、三聚体等)和肽或蛋白质之间残基间接触频率的Python脚本下载地址:
https://github.com/strodel-group/Oligomerization-State_and_Contact-Map
上述脚本是自动化运行的,仅仅需要protein_only.pdb和protein_nopbc.trr作为输入文件从而生成以下输出:
#Inputfiles protein_only.pdb protein_nopbc.trr
#Importantoutput files Oligomer_groups.dat:将相互作用的蛋白质链分组。Oligomer_states.dat:计算每个寡聚体组中的链数。Oligo-highest-size.dat:查找每个MD帧形成的最大寡聚体尺寸。Oligo-block-average.dat:创建最大寡聚体尺寸的25帧移动平均值。Contact_map.dat:保存不同蛋白质残基之间的接触频率。
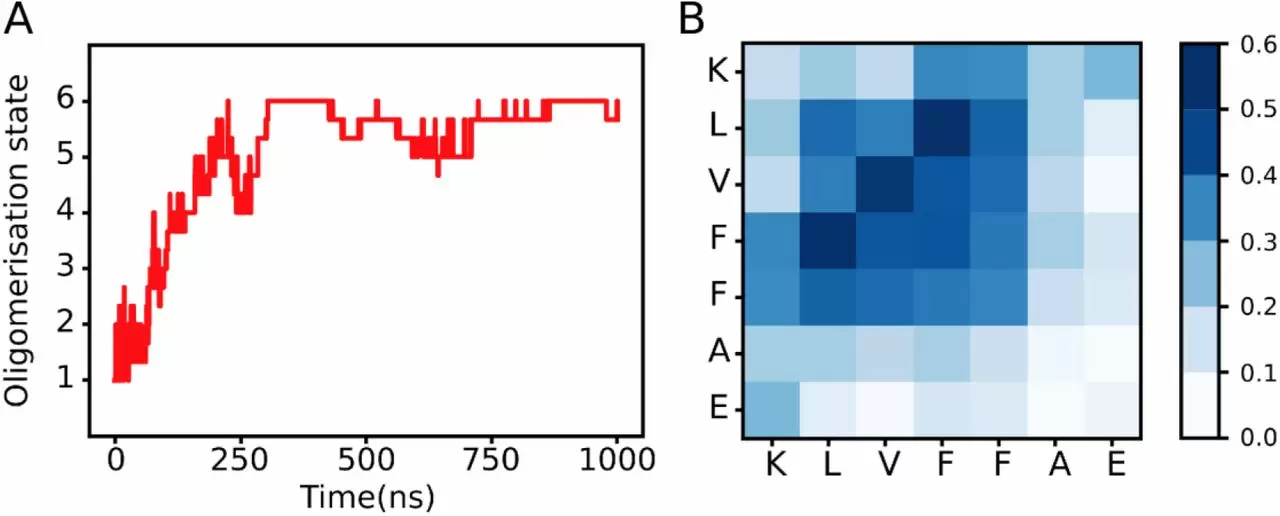
图2,六种Aβ16-22肽聚集后的1-μsMD 模拟分析。A)显示了系统随时间的低聚状态。B)显示了根据右侧色标的概率的残基间接触图。报告了模拟期间采样的组成寡聚体的肽之间的接触。
从应用于我们的示例分析中获得的结果如图2所示。图A报告了系统的寡聚化状态,如果相对于任何两个原子的最小距离低于0.4nm则两个蛋白质被认为是相互接触的。例如,如果系统在某一时刻由二聚体和四聚体组成,那么作为较大寡聚体的四聚体是有意义的。上述结果显示六种肽在300ns 内达到六聚体状态,但随后观察到一些解离和重新结合事件,特别是在500到750ns 之间。随后,图2B则显示寡聚体的肽之间的残基-残基接触的概率。它表明肽更倾向于以反平行方向组装,这通过带相反电荷的N端K16和C端E22残基之间的静电相互作用来稳定。此外,我们也发现其形成了一些强疏水接触,特别是Li17-Fj19、Vi18-Vj18和Fi19-Lj17之间(i和j分别指的是寡聚体中两个不同的肽段)。
总之,该文章提供了具体的操作步骤指南和必要的分析脚本文件,用于使用GROMACS 运行肽聚集的MD模拟,并利用寡聚体大小、肽间接触、马尔可夫状态模型(略)和过渡网络(略)分析了这些模拟数据。肽和蛋白质聚集与许多疾病有关,例如阿尔茨海默病和帕金森病。对于计算模拟,尤其是在微秒时间尺度上具有原子分辨率的MD模拟已成为研究肽或蛋白质的序列、构象特性和聚集之间关系的重要工具。由于MD模拟产生大量数据,因此开发从MD轨迹中提取信息的工具非常重要,这些信息可为所研究的过程提供关键见解。在研究聚集方面,关键问题在于模拟过程中是否形成了寡聚体,它们大小如何,以及聚集过程是通过什么相互作用来驱动的。这些问题可以通过计算寡聚体大小和寡聚体内的残基间接触来回答。另外,为了深入了解聚合路径,需要从MD数据中推断出适当的网络模型。我们提出了计算这种网络模型的两种可能选择,马尔可夫状态模型和过渡网络方法。前者基于MD数据的动力学聚类,阐述对动力学相关聚集途径的关键见解,如本文所证明的Aβ16-22肽的二聚化。另一方面,过渡网络(TN)则基于构象聚类,从而提供有关在聚合过程中采样的不同低聚物的更多结构细节。此外,用户也可以轻松控在TN中呈现的细节数目,譬如可以以寡聚体大小作为唯一描述符的粗粒度TN到每个状态具有多个描述符以区分低聚物的非常细粒度的TN。因此,MSM和TN分析可以相辅相成,但需要注意的是,MSM需要收敛的MD数据,这通常意味着几十微秒的MD 采样,否则将无法构建。
综上,本文通过寡聚体大小、接触图、MSM和TN可以分析研究肽或蛋白质聚集的MD轨迹,并为实现上述分析提供了必要的文件和说明。
原始文献:
Samantray S, Schumann W, Illig A M, et al. Molecular dynamics simulations of protein aggregation: protocols for simulation setup and analysis with Markov state models and transition networks[J]. bioRxiv, 2020.

 我的购物车
我的购物车
 粤公网安备 44030402005306号
粤公网安备 44030402005306号



