随着小分子策略方法的进步,药物研发成功率显著提高。因此,如今药物设计策略的重要目标已变成如何快速释放目标靶点的治疗潜力。事实上大量案例研究表明,快速辨别具有风险平衡的临床候选药物将显著推动靶点有效性评估。因此,我们需要强调临床经验在该过程中的决定性作用,从而确保通过高度可信的靶点提高药物研发的成功几率。
成功的小分子药物设计需要具有内在治疗潜力的分子靶点和具有正确特性的分子来释放其治疗潜能。当今的药物设计策略已经发展到几乎没有进一步改进类药特性空间。因此,与靶点相关的安全性或有效性不足如今已构成了II期临床前开发中损耗的主要原因。这一发现导致人们不得不更加关注于前期靶点的选择。事实上,能够快速识别风险平衡的临床候选药物,并将临床经验转化为有意义的差异化策略以及扩展可药用蛋白质组的设计策略是药物设计者可以加速发现下一代药物的重要手段。
二期临床试验是转化药物能够流向患者过程中最具挑战性的关键环节,因此降低该环节的高损耗率是制药行业面临的最重要和最困难的挑战。其成功取决于分子靶点是否确实具有治疗潜力以及候选分子能否真正彻底释放这种潜力,两者的结合在很大程度上决定了二期临床试验阶段成功的可能性。历史上导致药代动力学(PK)和安全性问题的次优分子特性一直是药物设计解决方案的损耗和机会的主要来源。因此,best-in-class设计策略可以改善first-in-class药物特性从而为患者带来更大价值。其中最好的例子来自于通过first-in-class药物硝苯地平的药物特性优化设计出best-in-class钙通道阻滞剂氨氯地平。加入碱性氮,增加分布容积而不对清除产生不利影响,创造了一种适合每天一次给药的分子,提高依从性,并提供更好的血压控制的同时减少了不良事件。然而,近几十年来,基于对最佳分子特性的理解和设计的相关原则的取得了重要的实质性进展。因此,当前first-in-class药物经常出现几乎已经是最优方案且没有任何改进的余地,即与小分子质量相关的问题已成为从临床前到二期临床阶段开发中相关损耗的一个相对次要的因素。2015年至2019 年的5年间,辉瑞(Pfizer)公司口服小分子药物的损耗分析支持了上述观点(图1a)。在这队列中,与分子靶点相关的问题构成了最大的损耗来源(47%),其中大部分与疗效不足有关(图1b)。相比之下,与分子质量相关的问题仅占整体损耗的19%,其中大部分与安全问题有关(图1d)并在成本最低的临床前开发阶段即可以快速解决(图1c)。与上述发现一致,过去20年的几项回顾性分析表明,与分子的PK特性(例如吸收、分布和清除)相关的问题相对于与功效和安全性相关的问题而言,是一个相对较小的损耗来源。迄今为止,还没有其他回顾性损耗分析能够完全清楚地区分哪些是分子自身问题引起的损耗以及哪些是基于靶点的损耗。据阿斯利康研究报告,在的后期成本更高的阶段,基于靶点的损耗风险更高,并且在靶点信心历来被认为是最低的(例如神经科学和肿瘤学)疾病领域,其总体损耗风险就特别高,进一步表明,在开发的各个阶段中,用于证明靶点有效性的二期临床成功率最低,也对总体研发成功率最为敏感。

图1. 2015 年至2019年口服小分子药物流失的回顾性分析。(a)完成临床前开发、I期或II 期的43 个项目的损耗来源。(b) 基于靶点的损耗来源。(c)按发展阶段划分的损耗来源。(d) 基于分子的损耗来源。基于靶点的损耗被定义为尽管有充分的靶点参与证据,但由于对有效性或安全性失去信心而导致的损耗。基于分子的损耗定义为由于候选分子的药代动力学或安全性不足而导致的损耗。“疾病区 (DA)退出”定义为相关治疗区撤资导致的损耗。Behind-in-class表示在没有令人信服的较优的差异化的情况下而导致的损耗。
因此,这些研究结果表明,旨在改善first-in-class分子的分子特性的设计策略不再代表提高药物发现项目组合生产力的转型杠杆。相反,当今first-in-class设计策略应该通过提高新分子靶点的临床评估效率来解决后期、基于靶点的损耗的首要风险。这样做有望加速实现患者价值,并且在某些情况下,可以提供临床相关的见解,这些见解可用于合理设计有意义的差异化后续分子。为此,当今的药物设计者还必须拓展新的技能,从而能够鉴别最有希望的分子靶点,其中许多靶点曾被认为是“不可成药的”。
鉴于二期临床所面临的挑战,能够在临床开发早期以耐受良好和基于适宜的开发剂量予以快速测试临床效果已成为提高研发成功率的关键部分。此外,与潜在候选分子相关的风险必须相对于其他总体风险(例如靶点疗效)进行考虑。
另据研究表明,首次合成的化合物与临床开发候选药物之间相当漫长,人们有足够充分的机会来提高候选分子的质量和识别效率。事实上存在许多途径可以快速发现和推进临床候选分子,包括高通量模拟合成;早期投入用于临床试验和监管毒理学研究的药物供应;选择具有易于扩展的化学结构以及对探索性毒理学研究的早期介入等等。然而大量经验表明,推动项目整体的进度的最大动力可能是在早期就在可开发的剂量上评估临床耐受性表现良好的小分子。这样的背景信息必然会有利于后期进一步优化药理学、毒理学和PK特性的必要平衡。为此,描述实现“类药”分子所需的分子特性平衡的经验指标有效地指导了设计策略。这些指标(例如LipE 和LipMetE)为分子设计指明了方向,并描绘了以前与现存药物相关的分子的三维空间特性(例如,用于口服ADME 特性的Ro5)。然而,最近的分析表明,分子特性空间的限制过于严苛,以至于严格遵守“类药空间”的相关规则可能会限制设计思路,特别是当我们考虑那些以前被认为是“不可成药”的分子靶标。因此,进一步描绘这种扩展的特性空间从而优化有助于口服有效药物分子的多参数方法将进一步高效鉴别各方面具有适当特性平衡的化合物。
另外,在优化先导化合物之前,遗传信息、患者概况和系统生物学越来越多地参与到PK 和PK/药效学(PK/PD)模型构建中,从而促进临床候选药物的有效设计、鉴定和开发。其中化学信息学、机器学习、人工智能和基于机制的转化建模方法一并助力于快速设计正确的临床候选分子。
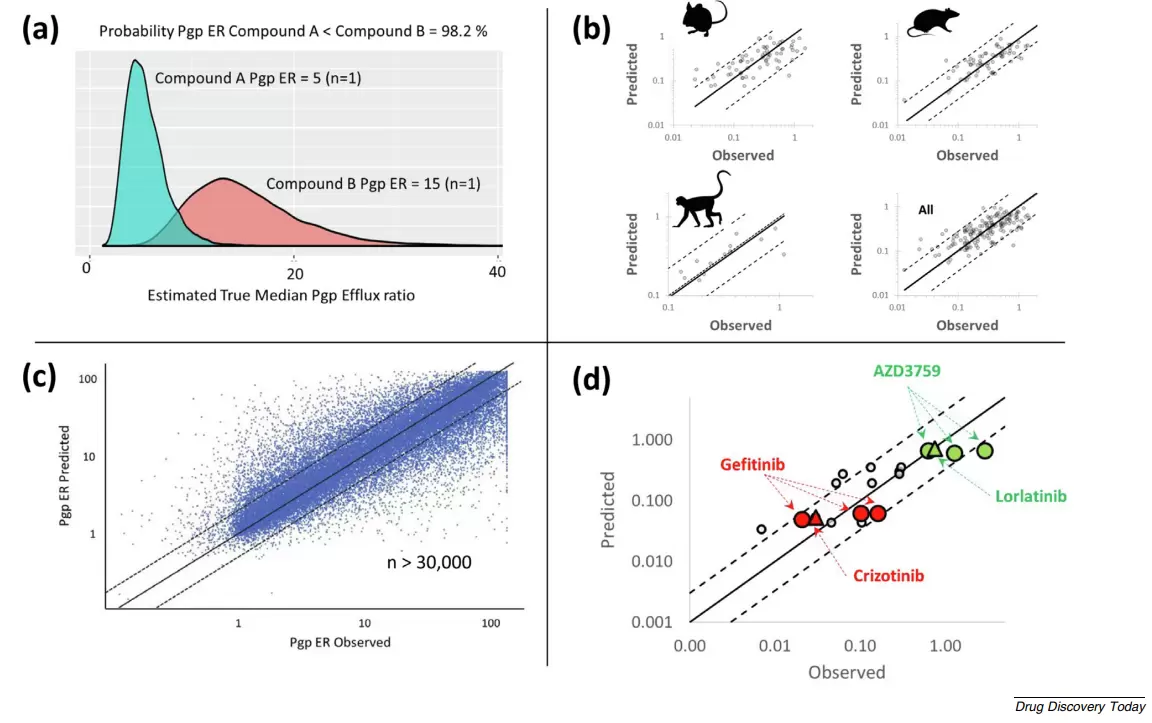
图2.早期描述用于设计血脑屏障渗透性化合物的转化平台。(a)高通量体外测定能够对化合物之间的外排活性进行特异性辨别。(b) 基于生理的药代动力学(PBPK)模型可用于跨物种的体外数据的体内转化过程。(c)基于机器学习模型可良好的预测不同化学结构的药物外排特征。(d)基于模型的人(三角形)及啮齿类动物(圆形)中一系列EGFR(N= 6)和ALK(N= 2)抑制剂的计血脑屏障渗透预测显示说明第一代(吉非替尼和克唑替尼)与之后设计用于改善渗透性的药物(AZD3759和劳拉替尼)明显的区分开来。
除了启用多参数先导优化策略之外,人工智能等也被用以加快药物发现过程,可涵盖从生物靶点的选择到先导识别以及化学合成等整个研发过程。其中一个值得关注的领域是开发更具预测性的模型用以从易于化学合成的超大型虚拟库中筛选出真正的目标分子。通过集成计算识别、先导优化和使用硬件技术合成等方法(如机器人、微流控等)提供了越来越自动化的药物设计平台。
药物安全性是成功的关键因素,提供具有足够治疗指数的分子设计策略对于快速识别临床候选化合物同样至关重要。鉴于毒性涉及的潜在机制,低剂量用药可能是确保安全的最普遍有效的手段。目前,基于先前数据和与结构或物理化学特性联系从而构建预测安全模型可能会加快安全的临床候选药物的设计与筛选。事实上,只要我们将对机制有充分的了解,就可以从毒理学发现转化为定量的“安全药理学”(例如QTc延长),从而建立具有可接受风险水平的安全范围。
最后,靶组织暴露毒性作用在药物设计策略中得到越来越多的认识和利用。通过结构设计以避免药物在末端毒性器官中的过度积累将有利于扩大治疗指数。类似的,通过克服药物分布的生理障碍或利用药物积累机制的策略也越来越多地利用组织内所需的作用部位来提高安全性(例如,通过应用纳米颗粒、结合转运蛋白等策略)。最后,药物靶点的差异表达也可以为设计具有更高安全性的药物提供另一种机会。
风险平衡的决策
在选择临床患者进行项目推进时应优先考虑那些有潜力为患者提供更具治疗意义的新治疗选择从而为之建立治疗优势。为此,设计人员应避免为找到具有“完美”特征的分子而造成时间延误,而忽略了机会风险的平衡。在许多情况下,即使是“不完美”的first-in-class分子也能为患者和创新公司提供价值。这一方面,布鲁顿酪氨酸激酶(BTK)抑制剂的研究提供了一个很好的例子。同类首创的共价抑制剂依鲁替尼是通过优化非选择性、非共价先导而发现的,并于2013年首次获得美国FDA批准用于治疗套细胞淋巴瘤,随后批准用于其他B细胞相关癌症,包括欧洲药品管理局(EMA) 和FDA 于2016年批准作为慢性淋巴细胞白血病的一线治疗药物。尽管依鲁替尼与不良反应相关,如血小板功能障碍、出血、心房颤动、腹泻、和皮疹,它通过替代耐受性较差的免疫抑制和细胞毒性疗法,并在后续药物治疗前数年为难治性/耐药性疾病患者提供更佳的选择,从而使患者切实获益。目前已有30多种共价和非共价BTK抑制剂已进入临床开发阶段,其中部分旨在通过更高的选择性改善患者在这些和其他适应症中的安全性和治疗体验,但迄今为止只有四种抑制剂获得批准(acalabrutinib、orelabrutinib、tirabrutinib和zanubrutinib)。目前看来,尽管整个行业进行了大量投资,并且有报道称后续药物具有更好的耐受性,但依鲁替尼仍然是目前的市场领导者,在这些后续药物多年之前就已为其批准的适应症患者提供了可接受的风险-收益。此例说明均衡风险及收益的重要性。
为了促进风险平衡的决策,有必要对基于分子的风险进行明确的评估。鉴于大多数基于化学分子的风险(例如剂量方案、安全性和药物-药物相互作用)只能根据疗效所需的暴露来理解,first-in-class药物在没有临床参照的前提下予以转化会带来更多的不确定性,但需避免过度努力在临床前鉴定完美分子。随着分子在开发阶段的进展,有关分子的风险与其有效浓度将不断调整,从而促进药物开发组织对项目的有效风险管理。
获得转化功效
旨在区分患者相关的差异化设计策略可以为不同类型的患者带来效果。通过药物靶点进行有意义的差异化设计策略最好通过先导分子的临床经验来了解,从而为创新公司提供竞争优势。此外,增加“druggable”蛋白质组的投入有望实现从遗传分析、表型筛选和其他受人类疾病影响的当代生物学方法中寻找最有希望的新靶点。
治疗差异化策略
毋庸置疑,有关先导分子以及first-in-class药物的临床经验不仅可以确定靶点的治疗潜力,而且还可以揭示限制实现全部治疗潜力的分子靶点的特征从而探索进一步研发策略。譬如一种用于治疗非小细胞肺癌(NSCLC)的第三代T790Mgatekeeper的EGFR抑制剂奥希替尼的研发。通过限制对血脑屏障的通过率使得第二代抗组胺药对中枢神经的镇静副作用显著降低。另外,一种第三代EML4-ALK抑制剂劳拉替尼显著提高了组织渗透性和抗耐药性突变的活性,在NSCLC中的疗效得到了显著改善。乙酰辅酶A-羧化酶抑制剂Clesacostat,通过其肝脏选择性分布而具有更高的安全性。
因此,为了达到既定目标而采取新的设计方法往往需要客观评估实现与患者差异化治疗策略,而不是渐进式改进从而导致后续分子,包括那些具有显著风险内部“备用”分子。
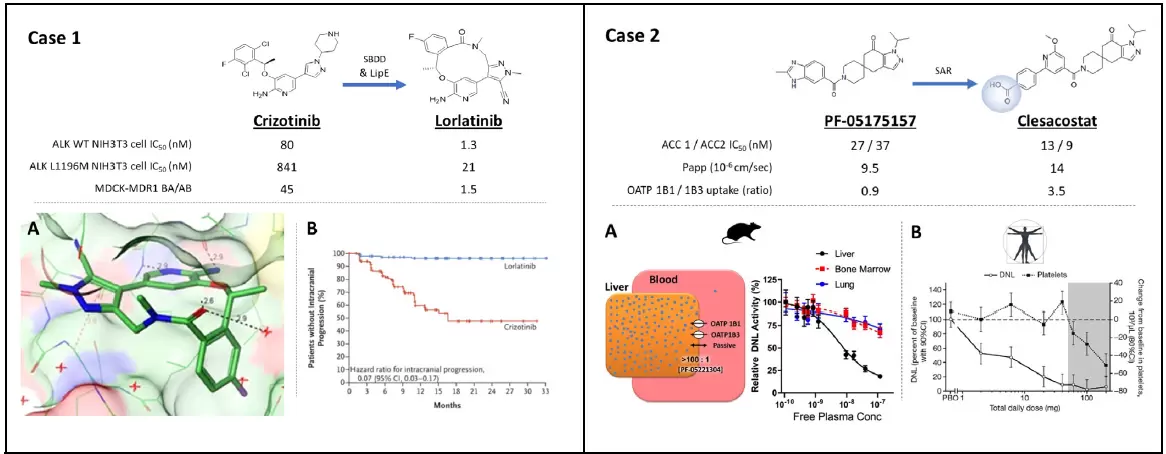
实例1,用于治疗EML4-ALK阳性非小细胞肺癌一线治疗药物lorlatinib的设计。A,该药物是基于first-in-classEML4-ALK抑制剂Crizotinib经过环化以及亲脂性、Pgp流出特性以及针对野生型与ALK突变型均具有良好活性的多参数优化而来。B,临床试验数据表明lorlatinib较crizotinib显著提高了患者的无进展生存率。
实例2,用于治疗NAFLD/NASH的乙酰辅酶A羧化酶抑制剂clesacostat(临床2b类药物)的设计。该化合物首先来自于一种可引起血小板减少症的ACC抑制剂,其主要原因是抑制了骨髓ACC活性。为此研究者将分子中引入羧基从而将药物选择性的经过肝脏OATP摄取达到了肝脏靶向性。A,临床前实验动物研究表明,cleascostat在小鼠肝脏富集。B,临床研究,在抑制肝脏脂肪生成作用的最大剂量下仍未发现clesacostat对血小板的抑制作用。
拓展“可成药”蛋白质组的新方法
关注新的靶点,包括历来被认为“不可抗药”的基因靶点,是不断征服人类疾病的基础。一系列创新方法被用于医药研发领域,如:1)筛选比传统高通量筛选(HTS) 更大的化合物集合(>109)(例如DNA编码文库技术);2)大型虚拟化合物库的计算筛选;3)新的(例如无标记)筛选方法;4)蛋白质上的变构结合位点的识别,这些位点可能提供活性位点无法提供的“成药性”机会;5)通过细胞、裂解物或蛋白质复合物筛选化合物,该方法比传统的生化分析更能反映其天然环境;6)筛选与大多数传统HTS筛选集合中的化合物不同的化合物,例如能够进行共价反应的化合物、大环化合物和天然产物。
其中最新例子包括一种获批用于治疗非小细胞肺癌的小分子KRAS G12C抑制剂sotorasib是通过共价优先方法发现的一隐秘结合口袋。另外,一种用于治疗糖尿病和肥胖症的B类GPCR,GLP-1R的小分子激动剂danuglipron源自一种在新型致敏高通量测定中具有活性的先导分子。对于通过与其他蛋白质或其复合物(例如骨架蛋白、分子伴侣或转录因子)相互作用影响多个细胞过程的靶蛋白存在特殊的挑战和机遇。在这些情况下,药物结合与涉及多种细胞过程相关的(例如增殖、分化、细胞凋亡、代谢、免疫反应、蛋白质转录、翻译、DNA修复)蛋白-蛋白相互作用使“成药性”变得更加复杂,如目前尚无批准的药物的c-MYC、p53和RAS以及尚未实现完全治疗潜力的临床证实的靶点,如雄激素和雌激素受体等。对于此类靶点,通过改变蛋白质表达水平(例如分子胶水、嵌合蛋白降解剂、蛋白质稳定剂、RNA调节剂等)以及调节蛋白-蛋白相互作用来扩大小分子的靶向范围。因此,有必要进行大量平行的研发投入从而了解和利用与此类新作用机制相关的安全性、有效性以及PK/PD的独特生物学特性,从而为患者提供更多的治疗策略。
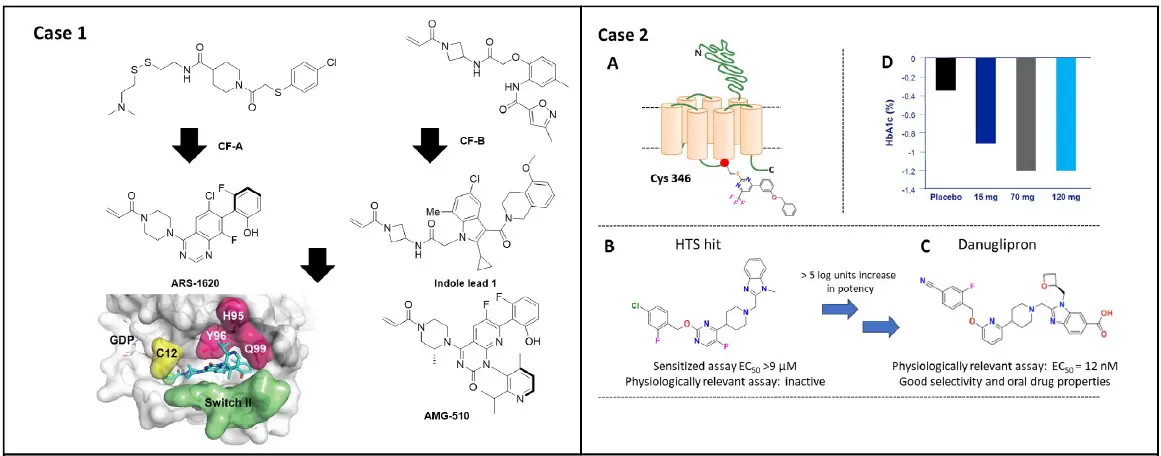
实例1,可口服的KRASG12C抑制剂AMG510的设计用于晚期结肠癌的治疗(二期临床)。Shokat与Amgen的的研究团队利用共价片断筛选技术获得了G12C抑制剂CF-A、CF-B。WellspringBiosciences的研究团队通过上述发现进一步基于结构药物设计出ARS-1610,同时Amgen团队设计出了IndoleLead 1。根据上述发现,尤其是IndoleLead 1诱导所引起的H95残基翻转从而发现一隐藏结合口袋,设计出一种强效高选择性KRASG12C抑制剂AMG510。
实例2,一种口服用于治疗2型糖尿病的的GLP-1R小分子激动剂danuglipron(PF-06882961)的设计发现。基于高通量筛选发现一较弱的GLP-1R激动剂(B),随后基于结构B优化设计出danuglipron(C),经4周治疗后该化合物表现出显著的降低2型糖尿病患者HbA1c水平,目前正处于2期临床。
除了基于靶点的干预方法外,表型筛选也是一种有希望的方法。鉴于表型筛选体现了更多的靶点空间。尽管该策略经常需要大量的下游研发投入以确保充分了解此类筛选的分子的作用机制和安全性,其潜在靶点和作用机制也常常不是那些事先被认为是“可成药靶点”(暗靶点)。最近批准的用于治疗囊性纤维化的药物,其中基于细胞表型筛选首先产生了CFTR 增强剂(如ivacaftor),然后CFTR 校正剂(如lumacaftor、texacaftor和elexacaftor),均是这类方法的例证。另外,一种抗HCV病毒NS5a抑制剂daclatasvir来自一种基于细胞的HCV复制子的筛选,该研究推动了进一步的NS5a抑制剂,如ledipasvir 和veltpasvir从而覆盖了其他HCV基因型。TYK2 抑制剂deucravacitinib(BMS-986165),最初的先导化合物是通过IL-23信号转导通路经过表型筛选发现的。随后的机制研究表明,该类化合物是TYK2变构抑制剂。目前正在开发用于治疗银屑病和其他自身免疫性疾病。
结束语
随着小分子药物设计技术的不断进步,其研发过程的分子损耗不断减少,其中基于靶点的损耗的在总体风险中占比较大,揭示了充分了解靶点的重要性。另外,虽然药物设计不能改变靶点的内在治疗潜力,但良好的设计策略可以充分发挥分子靶点的内在治疗潜力从而加快临床应用价值的实现,提高药物转化潜力。当前一系列研发实例表明,快速识别风险平衡的临床候选分子,并根据先导分子的临床经验制定一系列差异化策略,增加“可药物化”蛋白质组的投资是实现这一目标的有希望的策略。
参考文献:
T.S. Maurer et al., Drug Discovery Today (2021), https://doi.org/10.1016/j.drudis.2021.09.017

 我的购物车
我的购物车
 粤公网安备 44030402005306号
粤公网安备 44030402005306号



