合成致死现象由美国哥伦比亚大学遗传学家Calvin Bridges于1922年首次描述,他在一种常见的模式生物果蝇(Drosophila melanogaster)中注意到某些单基因的组合突变导致果蝇不能存活,而任何单基因突变却并不会造成果蝇的任何伤害。1946 年,同在哥伦比亚大学工作的Theodore Dobzhansky创造了“合成致死”一词来描述这种与果蝇类似的遗传相互作用。另外,作者也定义了一种称为“合成疾病”的概念,用以描述如果组合的遗传事件并不致死,而只是导致疾病发生这一现象。目前,合成致死的定义已超出了作者当初所描述的经典遗传学的范畴。譬如合成致死也适用于某一基因突变+另一基因功能采用化合物抑制的组合所导致的致死情况。
从机制上说,合成致死是生物体带有某种备份倾向的缓冲方案的结果。如某单一基因存在潜在的遗传变异、环境变化或其他随机事件(例如突变),由于在该过程存在备份选项,生物体仍具表型稳定性。这种所谓“平行冗余通路”保证了遗传上的稳定性。事实上人们发现,大多数重要的细胞过程并不依赖于任何单个组件。
合成致死可用于分子靶向癌症治疗。其原理是:如肿瘤细胞中存在某一特定基因A发生突变失活,此时细胞仅依靠其备选方案基因B存活,那么用药物等手段抑制基因B,使两者都失活而发生致死的现象。由于健康个体有正常的基因A,从而保证正常的生理功能不会受到伤害。
合成致死靶点的发现
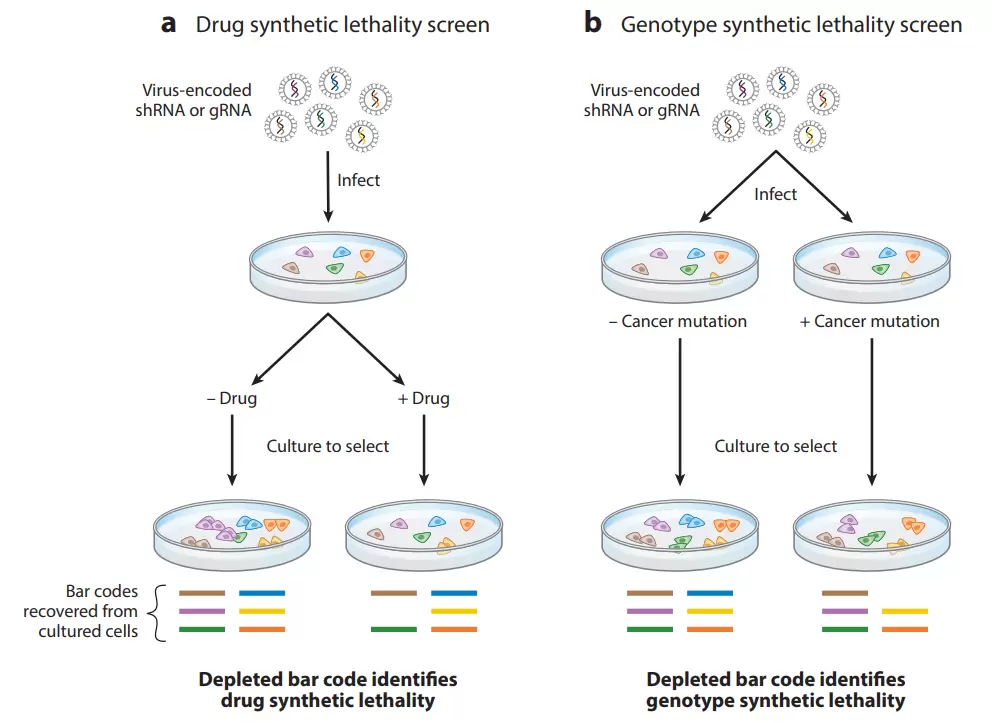
合成致死相互作用可以利用相关实验来发现、确定。目前,在特定的 基因型背景下,可以干扰全基因组中任一基因表达进而导致合成致死的表型筛选技术。其中包括大规模合成的短干扰RNA(siRNA)片段文库、短发夹RNA(shRNA)文库,以及最近用于CRISPR/Cas9基因组编辑的大量guide RNA(gRNA)文库。所有这些技术都可以高通量筛选形式进行,其中每个基因及试剂
BRCA1/2 与 PARP 的合成致死效应
利用合成致死方法靶向治疗的第一个例子是通过靶向基因损伤修复(DDR)通路而产生的。其针对类型为BRCA1/2(基因A)存在突变的肿瘤,这种针对PARP(基因B)的抑制剂的治疗方法于2014年获得FDA批准。PARP抑制剂研究历史颇具曲折性。1963年Chambon在研究RNA聚合酶的过程中,无意间发现了一种具有DNA聚合活性的酶,即多聚ADP核糖聚合酶(PARP)。1971年J.BClark找到了一种可以抑制PARP活性的物质-烟酰胺。几年后Barbara Durkacz等人证明烟酰胺的类似物(3-氨基苯甲酰胺,3-AB)可以抑制DNA的修复,并能增强DNA损伤剂硫酸二甲酯的细胞毒性。然而,3-AB活性较低,只能在毫摩尔浓度下才能抑制PARP。不过这也极大地激发了科学家们的兴趣,他们不断地寻找活性更高、选择性更强的PARP抑制剂。
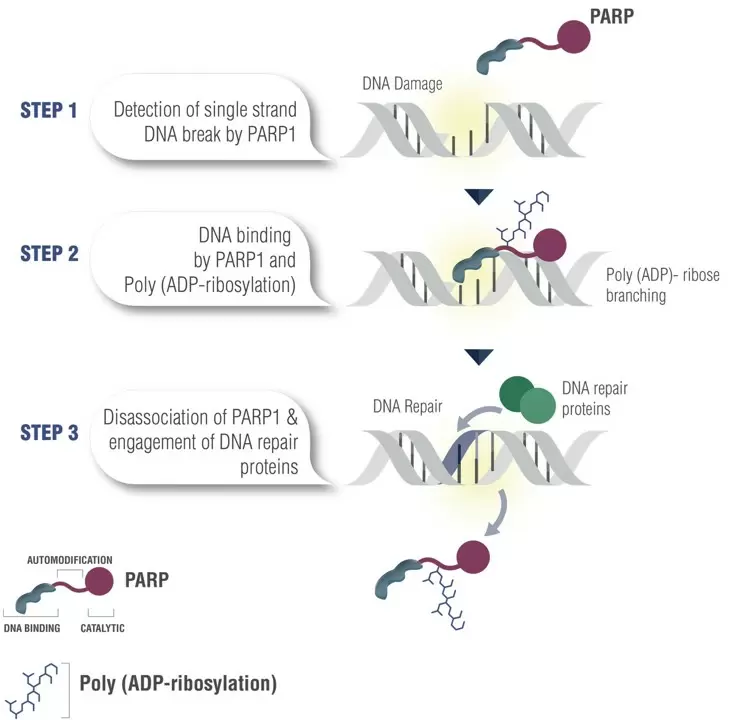
与此同时,人们发现存在17种PARP亚型,其中PARP1在DNA单链断裂修复中发挥关键作用。PARP1可以准确识别并结合DNA单链断裂的缺口,同时周围的烟酰胺腺嘌呤二核苷酸(NAD+)会迅速与PRAP-1的活性位点结合形成复合体,这个复合体会召集来其他参与DNA修复的效应分子,快速填补DNA断裂的缺口。在完成DNA修复后,PARP-1又从DNA上脱离下来,恢复至游离状态。
PARP的DNA损伤修复过程 (Gourley,et al., J Clin Oncol 2019)
除此之外,PARP家族中PARP2也能准确识别单链断裂,而对于同样在修复DNA损伤中发挥作用的PARP3,科学家们还没有彻底搞清其具体机制。
随后进一步研究发现,PARP1敲除的小鼠发育正常,但其姐妹染色单体交换水平显著增加。意味着PARP的缺失导致DNA增加了同源重组修复进程。DNA同源重组是修复DNA双链断裂的另一种方式,参与这种修复方式的蛋白有很多,其中最为人所熟知的是BRCA蛋白。
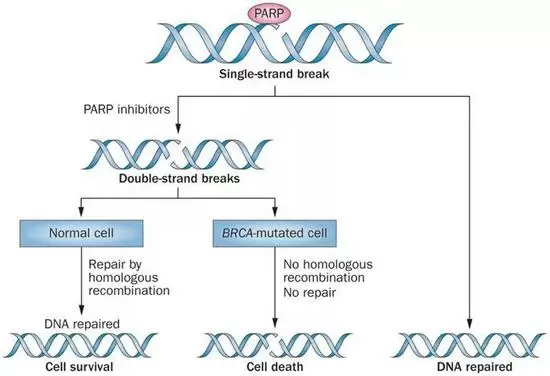
2005年英国的两个研究团队在同一期《自然》发表文章证实PARP抑制剂与BRCA1或BRCA2突变之间存在“合成致死”的相互作用。在BRCA功能正常的细胞中,当PARP被抑制之后,细胞中的单链断裂持续增加,而一旦单链断裂遇上了复制叉,DNA损伤就会变成双链断裂。此时,BRCA参与的同源重组就会担负起修复双链断裂的重任,确保DNA的完整性。
PARP 抑制剂与 BRCA“ 合成致死”
然而,在BRCA突变的癌细胞中,同源重组无法进行,再加上PARP的功能受到了抑制,面对大量的双链断裂,癌细胞只能选择一种快速却非常容易出错的非同源末端连接(NHEJ),导致细胞大量死亡。基于此“合成致死”理论,此后一大批PARP抑制剂奔赴临床试验。比如AZD2281,也就是后来的奥拉帕利(olaparib)。在临床研究中发现携带BRCA突变的患者群组中,奥拉帕利的PFS延长了6.9个月(11.2个月vs.4.3个月)在经过3期临床试验后,奥拉帕利在2014年先后获得欧洲药监局EMA和美国FDA上市批准,用于BRCA1/2突变的卵巢癌的治疗。
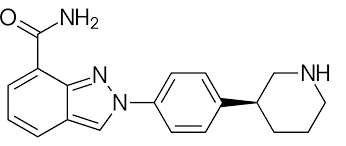
2009年,当芦卡帕利(rucaparib)、奥拉帕利都已经迈入临床研究之时,一个代号为MK-4827的化合物才第一次登上学术期刊,这就是后来的尼拉帕利(niraparib)。MK-4827是具有更高选择性的PARP1/2抑制活性,这也是其能后来居上的关键。
MK-4827(尼拉帕利)结构式
临床研究甚至发现,尼拉帕利同样能够使正常BRCA表达的患者收益。这也意味着尼拉帕利的使用并不需要考虑BRCA是否存在突变。尼拉帕利在2017年3月迅速获得美国FDA的上市批准,用于经铂类化疗后肿瘤完全或部分缓解(完全或部分反应)的成人复发性上皮性卵巢癌、输卵管或原发性腹膜癌的维持治疗。
尽管尼拉帕利比奥拉帕利和芦卡帕利,上市时间晚了几年,但这依然不妨碍尼拉帕利成为闪耀的新星,因为它是首个无需考虑BRCA突变或同源重组缺陷,就可用于治疗的PARP抑制剂。在其他癌症的联合治疗中,比如三阴性乳腺癌,一项Ⅱ期临床试验显示,尼拉帕利联合PD-1单抗(pembrolizumab)治疗的客观缓解率达到了29%,高于pembrolizumab单独治疗5–18%的客观缓解率,患者显著获益;此外,针对非小细胞肺癌、前列腺癌,PARP抑制剂联合化疗或放疗治疗的临床试验正在开展中。除了癌症之外,PARP抑制剂在心肌梗死、中风、慢性神经炎等也表现出很大的潜力。
TP53 与 WEE1 合成致死效应
TP53是人类非常重要的抑癌基因,TP53基因翻译的P53蛋白是细胞生长、增殖和损伤修复的重要调节因子。细胞的DNA受损时,P53蛋白可阻止细胞增殖 WEE1 则是G2期DNA 损伤修复通路相关的一个重要激酶。WEE1激酶能磷酸化细胞分裂周期基因(cell division cycle,CDC2),从而下调其活性,调控细胞G2到M期的转变并调节细胞有丝分裂。因此,WEE1与TP53在DNA损伤修复种存在一定的合成致死效应。另外从机制上看,凡能调控G2-M期的因子均能作为P53的合成致死基因。如ATM/ATR、CHK1/2、p38/MK2通路以及代谢通路中己糖激酶2(HK2)等。而另一方面,WEE1抑制剂亦可与PARP抑制剂或与化疗药物协同作用达到相互增敏的疗效。
RAS 合成致死基因的探索
尽管目前已有基于KRASG12C 突变药物的发现及临床应用,由于其与底物的高亲和等特点,RAS仍然是一种极难成药的靶点。因此,人们将大量注意力集中在其下游靶点,如MAPK和PI3K通路,包括RAF、MEK和ERK蛋白激酶,以及PI3K、AKT激酶和mTOR。尽管这些药物或其组合出现在许多临床试验中,但从数据分析这些治疗策略似乎只能提高无进展生存期(PFS)而不是总生存期(OS)。
目前,人们已经开展了几种遗传筛选来鉴定与RAS突变相关的合成致死基因,包括PLK1、STK33、TBK1、PKCδ、GATA2、TAK1和CDK1等。这些研究发现,RAS突变的细胞对有丝分裂的扰动特别敏感,当有丝分裂受到抑制时,它们会导致细胞在分裂前中期积累并死亡。然而值得注意的是,当使用反向筛选,即筛选对蛋白酶体或PLK1抑制剂的敏感基因时,却没有发现KRAS突变的明确相关性。因此基于这一筛选策略所获得的候选基因可能很大程度上与筛选细胞系相关、与特定的处理过程相关,亦有可能与不同的小分子抑制剂有关。事实上迄今并没有一种基于此策略发现的合成致死基因得到临床证实。
Myc 合成致死基因的探索
与RAS类似,Myc同样是一种较难成药的靶点。早期人们发现TRAIL死亡受体DR5激动剂可诱导Myc过表达细胞凋亡。其潜在机制是Myc可上调DR5的表达,从而增强了DR5激动剂对细胞的凋亡诱导作用。同样,在Myc过表达TRAIL诱导细胞凋亡增强剂的筛选中发现沉默GSK3β可阻止Myc在残基苏氨酸58上的磷酸化,从而抑制E3泛素连接酶FBW7对Myc的识别和随后的降解。上调的Myc导致DR5水平增加和DR5诱导的细胞凋亡的增强。该结果表明进一步增加Myc表达或者活性,可导致其第二种药物表现出更高的敏感性。另外,BRD4、AuroraA、SAE1、SAE2、ARK5、BUD31、SF3B1以及U2AF1等均被发现为Myc的合成致死基因。
RB1 合成致死基因的探索
与TP53类似,RB1属于抑癌基因,在肿瘤细胞通常属于突变或者表达失活状态。人们发现小分子CDK4/6抑制剂palbociclib可重新活化野生型RB1蛋白的表达。由于RB1蛋白突变的肿瘤并未有SOD2过表达,进而对ROS极为敏感,因此TSC2作为其中一种潜在的合成致死靶点可诱导一系列细胞代谢
附带致死( collateral lethality )作用
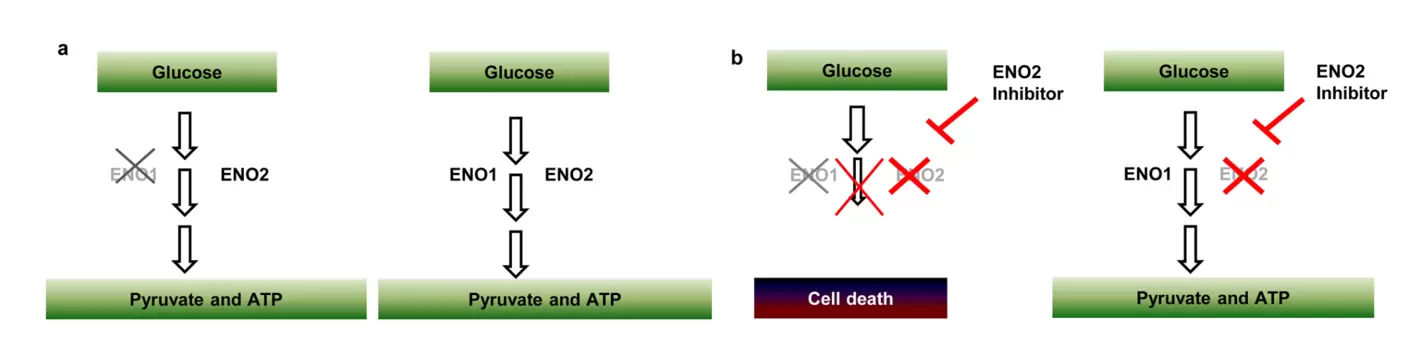
现实中人们发现另一个有趣的例子,即肿瘤细胞在同源删除抑癌基因的同时亦可能删除或影响位于临近的相关基因(称之为乘客基因)从而暴露其脆弱性,这就是所谓的“附带致死(collateral lethality)或者附带脆弱性(collateral vulnerability)”。例如,某些胶质母细胞瘤(GBM)中染色体1p36基因座中的糖酵解基因烯醇化酶1(ENO1) 被“附带删除”,此时ENO2的表达可以发挥代偿作用。 此时如干扰ENO2 活性则能抑制GBM 细胞的生长、存活及致瘤特性。另外使用烯醇化酶抑制剂膦酰乙酰异羟肟酸(PhAH) 亦能对ENO1 缺失的GBM细胞具有选择毒性但并不影响正常星形胶质细胞的存活。因此,“附带脆弱性”原则应适用于其编码的具有功能冗余但为生命代谢过程种至关重要的乘客删除基因,该现象为包含此类基因组事件的癌症提供了另一种潜在的治疗策略。
参考文献:
Roderick L.Beijersbergen, Lodewyk F.A. Wessels, and René Bernards SyntheticLethality in Cancer Therapeutics. Annual Review of Cancer Biology2017 1:1, 141-161

 我的购物车
我的购物车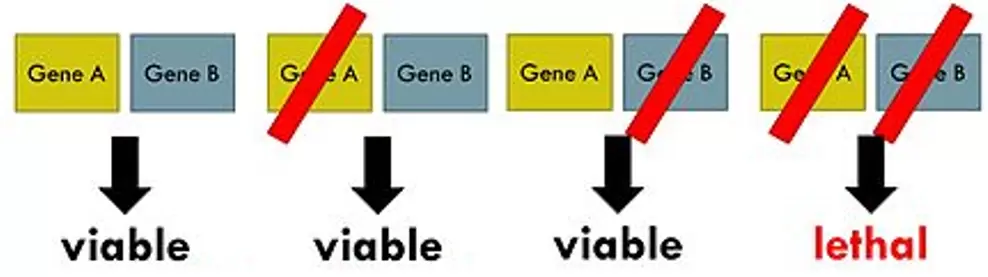

 粤公网安备 44030402005306号
粤公网安备 44030402005306号



